
Deep groove ball bearing 6311-skf 55x120x29 mm non-tight - 6311 bearing
Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB–an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and densitydependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Restricted open-shell DFT (RODFT) computations on geometries and FMOs of CH3 through CF3 are shown in Fig. 4. These RODFT calculations involve restricting all orbitals to be doubly occupied with an α and β electron, or to be vacant. Only one orbital is occupied by the radical electron, which simultaneously represents both the α-occupied orbital and the β vacant orbital. This differs from an unrestricted calculation, where α and β electrons occupy separate orbitals. The unrestricted DFT results that we have also performed are similar to RODFT but more complicated to describe, due to the presence of distinct sets of both α and β orbitals. In such cases, two different SOMO energies arise, depending on whether the SOMO is occupied or vacant.
Kino, T. et al. Trifluoromethylation of various aromatic compounds by CF3I in the presence of Fe(II) compound, H2O2 and dimethylsulfoxide. J. Fluor. Chem. 131, 98–105 (2010).
To provide a clearer explanation, we describe a simple model to explain the radical orbital energy in terms of F inductive withdrawal and conjugative donation (Fig. 7). In this model, the inductive effect of the fluorine is assumed to be approximately constant, as suggested by charges in Fig. 4c. Our calculations reveal that each subsequent fluorine substitution uniformly increases the charge on the carbon by a consistent value (0.13). Consequently, we presume that the substitution of each additional fluorine reduces the SOMO energy by an identical amount, here assumed to be −0.8 eV (purple numbers). This competes with conjugative electron donation by F, which raises the energy of the SOMO. As noted in Fig. 7, the conjugative effect (green numbers) somewhat exceeds the inductive effect for one F (0.9 eV) but diminishes for the second (0.5 eV) and third (0.2 eV) fluorines. Therefore, the SOMO energies of CH2F and CF2H are similar to CH3, due to near cancellation of conjugative donation and inductive withdrawal. These three radicals are nucleophilic. CF3 is electrophilic because of triple F inductive withdrawal and very weak π donation. The counteracting inductive and resonance effect of F is also cited in the perfluoro effect in photoelectron spectroscopy22, where, for example, perfluorination of benzene changes the HOMO energies (the first IP) very little.
Li, X., Zhang, S. Q., Xu, L. C. & Hong, X. Predicting regioselectivity in radical C-H functionalization of heterocycles through machine learning. Angew. Chem. Int. Ed. 59, 13253–13259 (2020).
De Vleeschouwer, F., Van Speybroeck, V., Waroquier, M., Geerlings, P. & De Proft, F. Electrophilicity and nucleophilicity index for radicals. Org. Lett. 9, 2721–2724 (2007).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
K.N.H. & P.S.B. conceived and directed the project. M.D. performed the DFT calculations. M.D., Q.S. & Q.Z. performed the data analysis. M.D., K.N.H. & P.S.B. wrote the paper with input from all other authors. All authors discussed the results and commented on the manuscript.
To demonstrate the utility of this model, regioselectivity differences between radical additions of CF3 and CF2H were investigated. For the nucleophilic CF2H radical, the interaction between the radical SOMO and the heterocycle LUMO is the most decisive. Consequently, the LUMO coefficients of the heterocycle dominate the regioselectivity. In the case of more electrophilic CF3 radical, the lower radical SOMO results in enhanced interaction with the heterocycle HOMO. Therefore, when dealing with electrophilic protonated heterocycles, both HOMO and LUMO coefficients have a significant effect on the regioselectivity of the CF3 radical addition. The difference between •CF3 and •CF2H reactivity and regioselectivity can be understood from these calculations and our FMO model.
All computations were performed using the Gaussian 1623. Geometry optimizations and frequency calculations in dichloromethane were conducted at the M06-2X/6-311 + G(d,p) level of theory with the SMD (Solvation Model Density) solvation model24,25,26,27,28,29. Following the geometry optimization, single-point energies and solvent effects in dichloromethane were calculated at the SMD-M06-2X/def2-QZVPP level of theory using the optimized structures30,31,32. Intrinsic Reaction Coordinate (IRC) calculations were carried out to confirm that the transition state connects the correct reactants and products.
a The energy profile and definition of ΔΔGTS. b Calculated relative Gibbs activation energies of transition states for the attack at C3, C5, or C7 as compared to the attack on C2 (ΔΔGTS). Computed ratios are given as black numbers, and experimental ratios of products are shown as gray numbers in parentheses. The results of the experiments are based on ref. 11.
Grimme, S. et al. Fully automated quantum-chemistry-based computation of spin-spincoupled nuclear magnetic resonance spectra. Angew. Chem. Int. Ed. 56, 14763–14769 (2017).
As depicted in Fig. 2b, in the first case of 4-acetylpyridine 1a, both CF2H and CF3 radicals favor attack at the C2 position. The more electrophilic CF3 radical shows a smaller preference for the C2 position compared to the CF2H radical (0.7 vs 1.1 kcal/mol), both experimentally and computationally. For both varenicline 1b and dihydroquinine 1c, the CF2H radical prefers the electron-poor position C2, while the CF3 radical favors the electron-rich positions C5 (−0.8 kcal/mol) and C7 (−1.3 kcal/mol). These predicted results show the same trends as the experimental observations. In addition, a study of unprotonated heterocycles with CF2H and CF3 radicals was conducted (Supplementary Fig. 1). The predicted results with unprotonated heterocycles do not match experimentally observed selectivity, supporting our surmise that protonation occurs under these reaction conditions. These findings are in good agreement with the Baran–Blackmond rules16.

Meanwell, N. A. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J. Med. Chem. 61, 5822–5880 (2018).
The initial phase of conformational exploration involved the use of Grimme’s CREST conformer-rotamer ensemble sampling tool, version 2.10.2 with xtb version 6.3.333,34,35,36. After the initial conformer generation, all the geometries below 5 kcal/mol were further refined using Gaussian 1623. The recalculations were conducted at the SMD-M06-2X/6-311 + G(d,p) level of theory, utilizing the Solvation Model Density (SMD) to simulate the influence of the solvent environment. This step was pivotal in accurately locating the lowest energy conformers.
Fluorinated and trifluoromethylated molecular scaffolds have extensive applications in the fields of agrochemicals, pharmaceuticals, and as materials1,2,3,4. Even though there are several methodologies for the fluoroalkylation of organic substrates, methods for achieving the direct difluoromethylation of heterocycles are underdeveloped5,6,7,8,9,10. Over a decade ago, zinc difluoromethanesulfinate (Zn(SO2CF2H)2, DFMS) was developed for direct difluoromethylation of heterocycles through a radical process11. This direct, scalable, user-friendly, and chemoselective difluoromethylation is compatible with a variety of organic substrates, including heterocycles, α,β-unsaturated enones, and aromatic thiols, finding extensive use in developing pharmaceuticals, agrochemicals, and other useful materials12,13.
All structures were confirmed as stationary points on the potential energy surface (PES) and characterized as either transition states or minima through frequency calculations. Specifically, transition states were identified by the presence of a single imaginary frequency in the vibrational analysis. Minima on the PES, representing stable structures such as reactants, intermediates, or products, were characterized by the absence of imaginary frequencies. Based on the optimized structures, normal vibrational mode analysis was carried out for all stationary points to derive the thermochemical corrections for the enthalpies and free energies.
Radical substitution is a useful method to functionalize heterocycles, as in the venerable Minisci reaction. Empirically observed regiochemistries indicate that the CF2H radical has a nucleophilic character similar to alkyl radicals, but the CF3 radical is electrophilic. While the difference between •CH3 and •CF3 is well understood, the reason that one and two Fs make little difference but the third has a large effect is puzzling. DFT calculations with M06-2X both reproduce experimental selectivities and also lead to an explanation of this difference. Theoretical methods reveal how the F inductive withdrawal and conjugative donation alter radical properties, but only CF3 becomes decidedly electrophilic toward heterocycles. Here, we show a simple model to explain the radical orbital energy trends and resulting nucleophilicity or electrophilicity of fluorinated radicals.
The predicted order for trifluoromethylation is according to the LUMO and HOMO densities. The experimental order is also shown.
Bernardi, F., Cherry, W., Shaik, S. & Epiotis, N. D. Structure of fluoromethyl radicals. conjugative and inductive effects. J. Am. Chem. Soc. 100, 1352–135 (1978).
With electrophilic protonated heterocycles, the SOMO-LUMO interaction dominates with a nucleophilic radical, and both SOMO-HOMO and SOMO-LUMO interactions are important with an electrophilic radical.
It’s well known that •CH3 is planar and relatively nucleophilic, while •CF3 is pyramidal and somewhat electrophilic17,18. The high electronegativity of F is countered by its ability to donate electrons by mixing of the F lone pair with the radical SOMO (Singly Occupied Molecular Orbital). As shown in Table 1, the IPs (Ionization Potential) of •CH3 through •CF3 gradually decrease with 1F or 2F substitutions, indicating dominance of lone-pair interaction. The IP then increases with 3F, suggesting electronegativity dominance. The absolute electronegativities (χ) show similar values for CH3, CH2F, and CF2H, but a considerably larger value for F. Similar trends are found in Parr’s theoretical index, global electrophilicity (ω)19. The computed SOMO energies also indicate that •CH3, •CH2F and •CF2H are similar, but only •CF3 is much more electrophilic. We show here why there is no gradual increase in electrophilicity with the substitution of 1 and 2 Fs, but only 3Fs make the radical electrophilic.
Grimme, S. Exploration of chemical compound, conformer, and reaction space with metadynamics simulations based on tight-binding quantum chemical calculations. J. Chem. Theory Comput. 15, 2847–2862 (2019).
Sap, J. B. et al. Late-stage difluoromethylation: concepts, developments and perspective. Chem. Soc. Rev. 50, 8214–8247 (2021).
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
Ogawa, Y., Tokunaga, E., Kobayashi, O., Hirai, K. & Shibata, N. Current contributions of organofluorine compounds to the agrochemical industry. iScience 23, 101467 (2020).
Dolbier, W. R. Jr, Médebielle, M. & Ait-Mohand, S. Single electron transfer approaches to the practical synthesis of aromatic and heterocyclic-CF2H derivatives. Tetrahedron Lett. 42, 4811–4814 (2001).
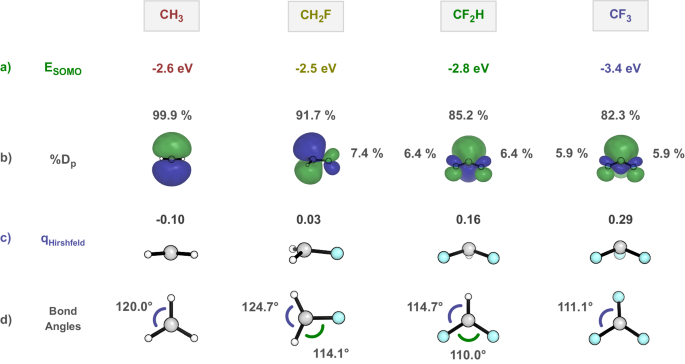
Returning to RODFT, Fig. 4 illustrates that the SOMO energies of CH3 (−2.6 eV), CH2F (−2.5 eV), CF2H (−2.8 eV), and CF3 (−3.4 eV) first increase in energy and then decrease, slightly with CF2H and dramatically with CF3. The percent orbital density decreases on the p orbital, by 8%, 7%, then only 3% in the series. The charge on the carbon becomes more positive along the series. Inspection of geometries indicates that there is slight pyramidalization of the radical with one fluorine, which becomes more pronounced with two and three fluorines (Fig. 4d). The pyramidalization is reflected in the deviation of the sum of the three angles at the radical center from 360° (the value for the planar methyl radical). These deviations from CH3 through CF3 are 0°, 7°, 21°, and 27°, a gradual pyramidalization not reflected in the abrupt difference in electrophilicity with 3Fs. There are only minor changes in the SOMO energies for CH2F and CF2H compared to CH3, whereas a large decrease in the SOMO energy with the addition of three fluorines. This indicates CF2H is similar to CH3, but CF3 is different.
The CF3 radical also has a more pyramidal structure than the CF2H radical, decreasing the degree of overlap between radical SOMO and fluorine π-type lone pair. This effect is small compared to the small coefficient and lower energy of the all-bonding combination of F lone pairs in CF320,21. The π donation and σ withdrawal by F is well known and discussed earlier in the references17,18,20. However, the earlier studies generally presented a smooth transition of nucleophilic through electrophilic behavior in methyl through trifluoromethyl. We emphasize why CH3 through CHF2 changes little in nucleophilic/electrophilic behavior, and only CF3 is different.
As noted earlier, fluorine substituents affect the radical SOMO energy in two ways, as depicted in Fig. 517,18. One is inductive; the fluorine is electronegative, lowering the energy of the SOMO orbital. The second effect is conjugative; fluorine is a π-donor, which leads to an increase in SOMO energy, due to the antibonding interaction of the radical SOMO with the lower-lying F lone pair. At the same time, the fluorine lone-pair orbitals are stabilized by mixing with higher-energy radical SOMO orbitals in a bonding fashion.
Berger, R., Resnati, G., Metrangolo, P., Weber, E. & Hulliger, J. Organic fluorine compounds: a great opportunity for enhanced materials properties. Chem. Soc. Rev. 40, 3496–3508 (2011).
Sang, Y., Zhou, B., Zheng, M. M., Xue, X. S., & Cheng, J. P. DFT modeling of catalytic fluorination reactions: mechanisms, reactivities, and selectivities. in Organofluorine Chemistry: Synthesis, Modeling, and Applications. 307–362 (Wiley-VCH, 2021).
Parsaee, F. et al. Radical philicity and its role in selective organic transformations. Nat. Rev. Chem. 5, 486–499 (2021).
Smith, J. M., Dixon, J. A., deGruyter, J. N. & Baran, P. S. Alkyl sulfinates: radical precursors enabling drug discovery: miniperspective. J. Med. Chem. 62, 2256–2264 (2019).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396 (2009).
We have described DFT calculations that elucidate why CF2H is a nucleophilic radical while CF3 is more electrophilic toward protonated heterocycles. To represent the F inductive withdrawal and conjugative donation, a simple model is proposed to explain the SOMO energies of CH3 through CF3. The analysis indicates that CH2F and CF2H have similar SOMO energies to CH3, due to the near cancellation of conjugative donation and inductive withdrawal. These are nucleophilic radicals. With three fluorines, the SOMO energy of CF3 is greatly lowered, because of triple F inductive withdrawal and weak π donation. This radical is relatively electrophilic.
Vleeschouwer, F. D., Speybroeck, V. V., Waroquier, M., Geerlings, P. & Proft, F. D. An intrinsic radical stability scale from the perspective of bond dissociation enthalpies: a companion to radical electrophilicities. J. Org. Chem. 73, 9109–9120 (2008).
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
To better clarify how fluorine π-type lone pair interacts with the radical SOMO, Fig. 6 presents a plot of different occupied orbitals, primarily p orbitals on fluorine, overlapping with the radical SOMO. A similar diagram is given in Bernardi et al.20. In the first case, the single fluorine lone pair mixes in a bonding fashion with the methyl SOMO orbital. In the second case, there are two fluorines, which form a bonding combination at −16.5 eV and an antibonding at −14.9 eV. However, only the −16.5 eV orbital has the right symmetry to mix with the radical SOMO orbital. As a result, the conjugative destabilization of the SOMO by antibonding interaction, and the orbital raising effect, is smaller with two fluorines than it is with one. And finally, with three fluorines, there are three different combinations of fluorine lone pairs. Only the one that involves bonding between all three fluorines, having the correct symmetry, can interact with the methyl SOMO. This orbital is much lower in energy and has smaller coefficients on fluorine, resulting in only a relatively minor destabilization of the SOMO.
a Restricted open-shell orbital energies of different radicals. b Side views of radical SOMO and percentages of orbital density at P in each SOMO (%Dp). c Side views of radicals and Hirshfeld Charges. d Top views of radicals and bond angles.
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Grimme, S., Bannwarth, C. & Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 13, 1989–2009 (2017).
As depicted in Fig. 9, for the heterocycle 4-acetylpyridine, both LUMO and HOMO coefficients are large at the C2 position. In the case of varenicline, the LUMO coefficients follow the order of C2 > C5 (20.2 vs. 5.8). By contrast, the order of HOMO coefficients is inverted (C5 » C2, 27.3 vs. 0.0). Thus, the final preference for varenicline is C5 over C2. Regarding dihydroquinine, the LUMO coefficients are ordered as C2 > C7 (18.7 vs. 10.3). Due to the node on C7 of the HOMO, the radical SOMO interacts with the dihydroquinine HOMO-1 instead of the HOMO (See Supplementary Fig. 4 for detailed explanation). The coefficients of HOMO-1 contribute to the positional differentiation. The HOMO-1 coefficients are arranged in the order: C7 » C2 (25.3 vs. 6.0). As a result, dihydroquinine shows a preference for C7 compared to C2. This explains the experimental observations.
Fujikawa, K., Fujioka, Y., Kobayashi, A. & Amii, H. A new method for aromatic difluoromethylation: copper-catalyzed cross-coupling and decarboxylation sequence from aryl iodides. Org. Lett. 13, 5560–5563 (2011).
Duan, M., Shao, Q., Zhou, Q. et al. Why •CF2H is nucleophilic but •CF3 is electrophilic in reactions with heterocycles. Nat Commun 15, 4630 (2024). https://doi.org/10.1038/s41467-024-48949-z
Yang, X., Wu, T., Phipps, R. J. & Toste, F. D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 115, 826–870 (2015).
Linstrom, P. J., & Mallard, W. G. NIST Chemistry WebBook, NIST Standard Reference Database Number 69 (National Institute of Standards and Technology, Gaithersburg, MD) https://doi.org/10.18434/T4D303.
Krishnan, R. B. J. S., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654 (1980).
Finally, we focus on the differences in regioselectivity between CF2H and CF3 radical additions. Following the earlier discussion (Fig. 3), for the nucleophilic CF2H radical, the radical SOMO mainly interacts with the protonated heterocycle LUMO. As a result, the SOMO-LUMO interaction will be the most decisive interaction for CF2H radical addition, and the LUMO coefficients of the protonated heterocycle dominate the regioselectivity. As presented in Fig. 8, the order of LUMO coefficients is C2 > C3 (12.9 vs. 7.0) for the simple heterocycle 4-acetylpyridine, C2 > C5 (20.2 vs. 5.8) for varenicline, and C2 > C7 (18.7 vs. 10.3) for dihydroquinine. This matches the experimentally observed regioselectivity.

In the case of the CF3 radical, the electrophilic CF3 radical SOMO has a lower orbital energy, which reduces the interaction of the SOMO with the protonated heterocycle LUMO. Thus, the positional differentiation brought by the protonated heterocycle LUMO will be less pronounced. The lower radical SOMO energy leads to an increase in SOMO-HOMO interaction. Therefore, both HOMO and LUMO coefficients contribute to determining the regioselectivity of the CF3 radical addition.
The DFT (Density Functional Theory) calculations were conducted first to explore the differences in regioselectivity found experimentally (Fig. 2). As the reaction conditions include TFA (trifluoroacetic acid) and H2O generally in mixed CH2Cl2/H2O solvent, we examine CF2H and CF3 radicals assuming protonated heterocycles (Fig. 2a). The transition states for attack of CF2H and CF3 radicals on different positions were computed. The calculated Gibbs activation energy differences (ΔΔGTS) account for the positional selectivity. Figure 2b depicts the calculated energy of the transition states for the attack at C3, C5, or C7 as compared to the attack on C2 (ΔΔGTS). Both computed ratios (black numbers), and experimental product ratios (gray numbers in parenthesis) are presented.
Schäfer, A., Huber, C. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 (1994).
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330 (2008).
Duan, M. et al. Chiral phosphoric acid catalyzed conversion of epoxides into thiiranes: mechanism, stereochemical model, and new catalyst design. Angew. Chem. Int. Ed. 61, e202113204 (2022).
All values are in eV. (−0.8 represents assumed constant inductive stabilization by each F; +0.9, +0.5, +0.2 represent assumed conjugative destabilization that decreases as the number of Fs increase).
We then focused on why CF3 is so much more electrophilic than CF2H, and why CF2H behaves like CH2F and CH3 as well as other alkyl and aryl radicals. As shown in Fig. 3, a simple Fukui frontier molecular orbital (FMO) model considers the interaction of the SOMO with the HOMO (Highest Occupied Molecular Orbital) and LUMO (Lowest Unoccupied Molecular Orbital) of the protonated heterocycles. There are three electron (SOMO-HOMO) and one electron (SOMO-LUMO) interactions, respectively, and both are stabilizing. One of these interactions may dominate. With electrophilic protonated heterocycles, a nucleophilic radical has a high-lying SOMO, and it interacts mainly with the heterocycle LUMO. In electrophilic radicals, the SOMO energy is lower, and the SOMO-HOMO interaction also becomes important. This pattern is because protonated heterocycles are intrinsically electrophilic. A more sophisticated analysis based on open-shell calculations and a GCDA (Generalized Charge Decomposition Analysis) method is given in the SI (Supplementary Fig. 2 and 3).
Nature Communications thanks Nand Kishor Gour, Rita Kakkar, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
To a solution of heterocycle (0.25 mmol, 1.0 equiv) and zinc difluoromethanesulfinate (DFMS) (200 mg, 0.50 mmol, 2.7 equiv) in dichloromethane (1.0 mL) and water (0.4 mL) at rt was added trifluoroacetic acid (20 µL, 0.25 mmol, 1.0 equiv) followed by slow addition of tert-butylhydroperoxide (70% solution in water, 0.17 mL, 1.25 mmol, 5.0 equiv) with vigorous stirring. The reaction was monitored by thin-layer chromatography until completion. For substrates that do not go to completion in 24 h, a second addition of DFMS (2.7 equiv) and tert-butylhydroperoxide (5.0 equiv) may be added to drive the reaction further. Upon consumption of the starting material, the reaction was partitioned between dichloromethane (2.0 mL) and saturated sodium bicarbonate (2.0 mL). The organic layer was separated, and the aqueous layer was extracted with dichloromethane (3 × 2.0 mL). The organic layers were dried with sodium sulfate, concentrated, and purified by column chromatography on silica gel.
The Natural Atomic Orbital (NAO) method was employed for an in-depth analysis of electronic structures37. This approach enables the calculation of orbital density percentages at each atom. To deepen understanding of the electronic interactions within and between molecules, the Multiwfn program was used to conduct the Generalized Charge Decomposition Analysis (GCDA) calculations38,39.
During these studies, it was observed empirically that the CF2H radical behaves much like alkyl and aryl radicals when added to heterocycles14,15,16. All possess nucleophilic character, reacting at the electrophilic regions of the heterocycle. However, when a single additional fluorine atom is added, the CF3 radical behaves as an electrophilic species. A comparison of CF2H and CF3 radical additions to varenicline and dihydroquinine demonstrates this (Fig. 1). In both cases, difluoromethylation takes place at electron-poor positions adjacent to nitrogen of the heterocycles (both at C2). This is found also with aryl and alkyl radicals14,15,16. In contrast, radical C–H trifluoromethylation occurs at the most electron-rich areas within the heterocycles (C5 and C7). Why is CF3 radical so much more electrophilic than CF2H, which behaves like alkyl radicals with these heterocycles? Computational studies are conducted to explore and elucidate this pronounced difference, offering a comprehensive explanation for the distinct reactivity. We describe how the geometries and orbital energies of different alkyl and fluorinated radicals show a distinct discontinuity between the difluoromethyl and trifluoromethyl radicals.
O’Hara, F., Blackmond, D. G. & Baran, P. S. Radical-based regioselective C−H functionalization of electron-deficient heteroarenes: scope, tunability, and predictability. J. Am. Chem. Soc. 135, 12122–12134 (2013).
Clark, I. D. & Frost, D. C. A study of the energy levels in benzene and some fluorobenzenes by photoelectron spectroscopy. J. Am. Chem. Soc. 89, 244–247 (1967).
Beatty, J. W., Douglas, J. J., Cole, K. P. & Stephenson, C. R. A scalable and operationally simple radical trifluoromethylation. Nat. Commun. 6, 7919 (2015).
We are grateful to the National Science Foundation (CHE-2153972 to K.N.H.) for the financial support of this research. Calculations were performed on the Hoffman2 cluster at UCLA, and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575 to K.N.H.).
To a solution of heterocycle (0.25 mmol, 1.0 equiv) and sodium trifluoromethylsulfinate (117 mg, 0.75 mmol, 3.0 equiv) in dichloromethane (1.0 mL) and water (0.4 mL) at 0 °C was slowly added tert-butylhydroperoxide (70% solution in water, 0.17 mL, 1.25 mmol, 5.0 equiv) with vigorous stirring. The reaction was allowed to warm to room temperature and monitored by thin-layer chromatography until completion. For substrates that do not go to completion in 24 h, a second addition of sodium trifluoromethylsulfinate (3.0 equiv) and tert-butylhydroperoxide (5.0 equiv) may be added to drive the reaction towards completion. Upon consumption of the starting material, the reaction was partitioned between dichloromethane (2.0 mL) and saturated sodium bicarbonate (2.0 mL). The organic layer was separated, and the aqueous layer was extracted with dichloromethane (3 × 2.0 mL). The organic layers were dried with sodium sulfate, concentrated, and purified by column chromatography on silica gel.
All data generated in this study are available in the Supplementary Information or from the corresponding author upon request. Source data containing the cartesian coordinates of optimized structures are provided. Source data are provided with this paper.
Weigend, F., Furche, F. & Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H-Kr. J. Chem. Phys. 119, 12753–12762 (2003).



 8613869596835
8613869596835